
Who We Are?
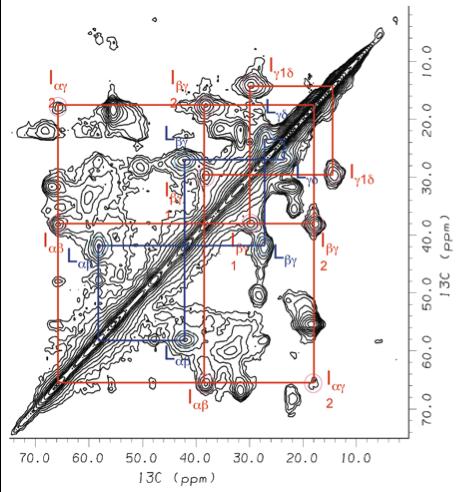
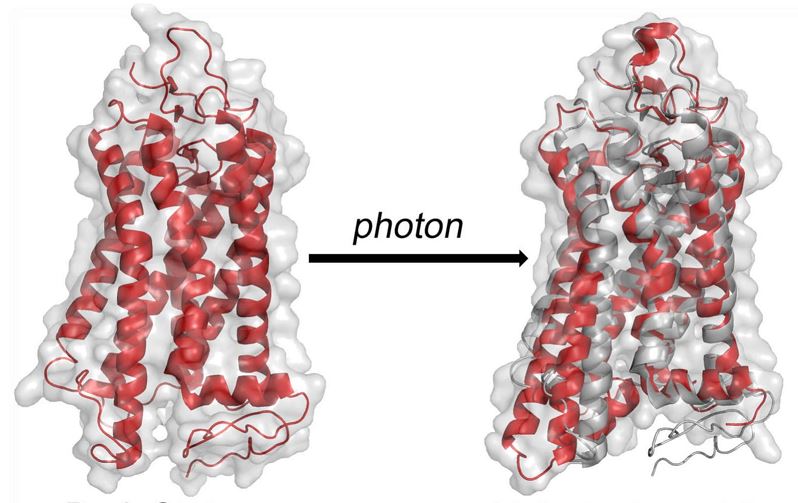
We are a group of Chemists, Biochemists and Physicists dedicated to structure and functional studies of biomolecules and biomembranes. We carry out fundamental research in molecular spectroscopy together with applications to membrane proteins, lipid bilayers, surfactants, and liquid crystals—soft matter in general.